
A Calculation of the Relative Risk of Pharmaceuticals Versus Nutrition Supplements
Executive Summary
- Health authorities point to the risks of nutrition supplements, without calculating the relative risk versus pharmaceuticals.
- We perform this calculation in this article.

Introduction
This article performs a calculation to compare the relative risk of pharmaceuticals to nutrition supplements.
Our References for This Article
If you want to see our references for this article and related Brightwork articles, visit this link.
Comparative Risk #2: The Issue of Adverse Reactions from Pharmaceuticals
The Relative Risk of Hospitalizations
The adverse reactions from pharmaceuticals overall are eye-popping.
Adverse drug reactions (ADR) are far more commonplace than one would think. It is estimated that ADRs represent the fourth leading cause of death in the United States and Canada behind heart disease, cancer, and stroke. Further, it is estimated that ADRs are the sixth leading cause of death worldwide.
Recent meta-analysis of prospective ADR studies estimates that over 180,000 Americans will die from ADRs and over one million will be injured from ADRs in 2008. Although these data are controversial and the actual incidence of ADRs is impossible to assess, there is no doubt that ADRs have a significant impact on both the healthcare delivery and the drug development industries.
Yet another way to demonstrate the impact of ADRs is to realize that approximately 5% of all hospital admissions are a direct result of ADRs, and unfortunately incidence has not changed over the past 30 years. – Science Direct
And from this quote.
Few know that systematic reviews of hospital charts found that even properly prescribed drugs (aside from misprescribing, overdosing, or self-prescribing) cause about 1.9 million hospitalizations a year. Another 840,000 hospitalized patients are given drugs that cause serious adverse reactions for a total of 2.74 million serious adverse drug reactions. – Harvard Ethics
By comparison, in the previous quote, the American Cancer Society pointed to 7,000 people taking supplements being either visiting health care facilities. And the New England Journal of Medicine article stated that while there were 23,000 visits to ERs, 2000 required hospitalization as the New England Journal of Medicine article specifies the number of hospitalizations, while the American Cancer Society does not, let us use the NEJM article’s estimate to perform some math of the comparative risk.
That is 1.7 M / 2,000 = 850x
What is interesting is that the CDC estimate of the number of patients hospitalized from ADRs is much lower than 1.7 M, as the following quotation states.
About 350,000 patients each year need to be hospitalized for further treatment after emergency visits for adverse drug events. – CDC
Everytime a health authority quotes a number with respect to ADRs, it is always lower than the values I find in an independent study. For this reason, I am disregarding this estimate from the CDC. It is well known at this point that the CDC is a puppet of drug companies.
That means that just using the overview estimates (excluding the suspicious CDC number). Pharmaceuticals are 850x more likely to result in hospitalization than supplements.
Is the American Cancer Society placing their emphasis in the right place? Furthermore, even this multiple is too low because to case nutrition supplements in the worst possible light. The medical establishment places many items, not nutrition supplements, into this category.
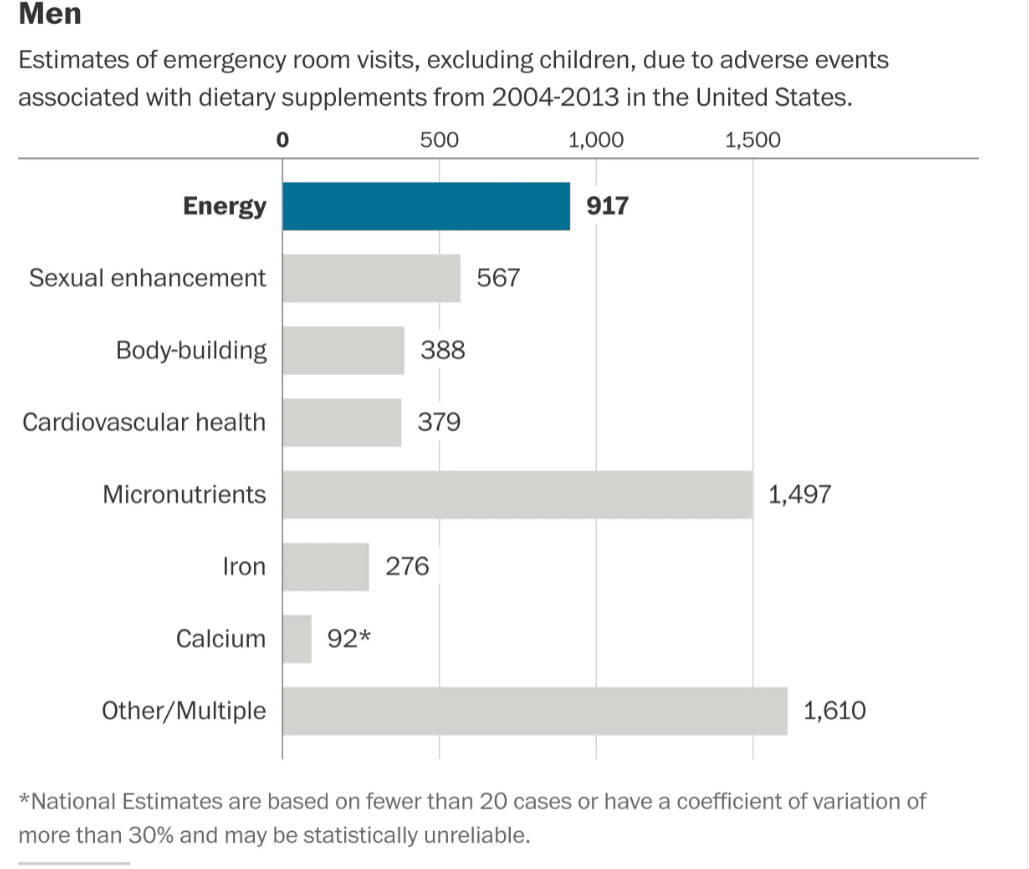
This is shown in the following graphic from the NEJM article.
Reactions for Men
Reactions for Women
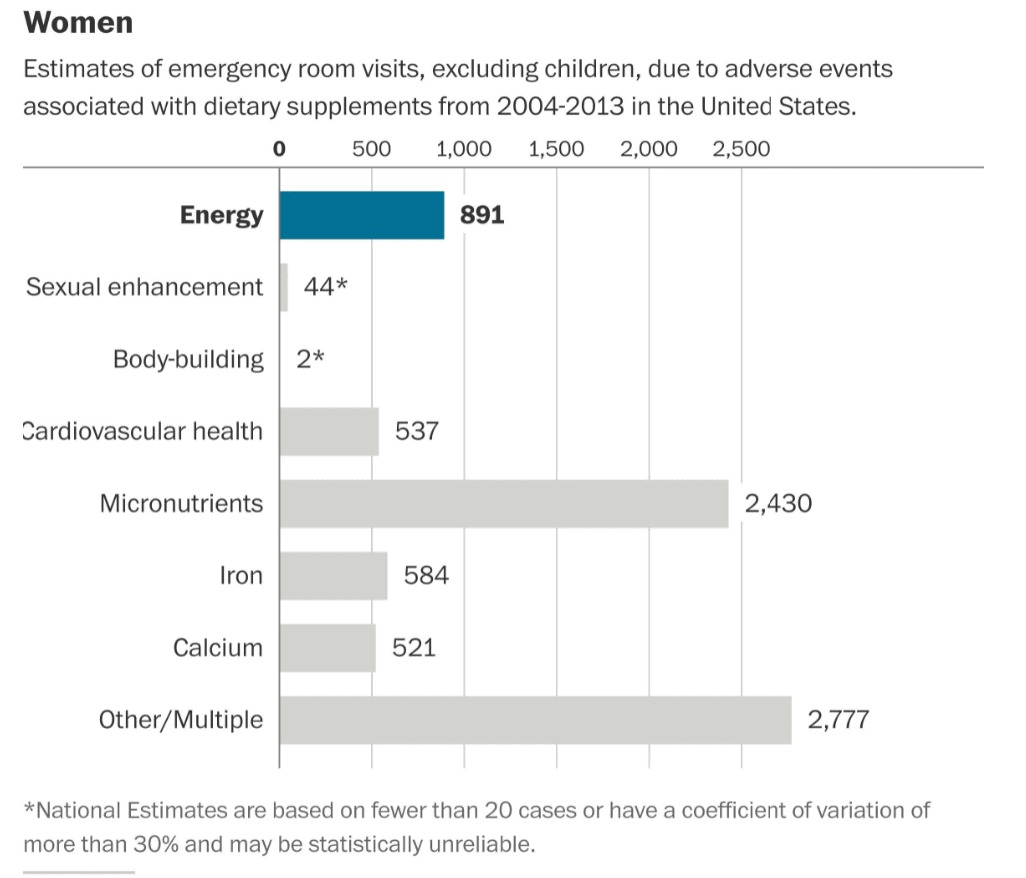
Graphical Source: Washington Post
Let us remove items that are not specifically nutrition supplements.
- No one should be taking energy drinks — and they are not nutrition supplements.
- Sexual enhancements are also not in the topic area. Some of the items under bodybuilding are not what I would consider nutrition supplements, and many of these people going to the ER may have had complications from steroid use. Steriods are not a nutrition supplement.
- Some of the items in “Other/Multiple” are things like eye drops.
Once some of these additions are removed, and only nutrition supplements are included, the risk of being hospitalized for a nutrition supplement declines to less than 1/1000x the risk of being hospitalized for pharmaceuticals.
The Relative Risk of Deaths
But this is only the risk for hospitalizations. That comparison is the best case scenario for pharmaceuticals vis-a-vis supplements. Once one switches to deaths, the relative risk moves even more in supplement’s favor.
When attempting to obtain an accurate estimate of the number of deaths per year from pharmaceuticals, we know that the FDA will provide the least accurate estimate, as they are part of the pharma complex. Here is the FDA’s estimate.
However, other studies conducted on hospitalized patient populations have placed much higher estimates on the overall incidence of serious ADRs. These studies estimate that 6.7% of hospitalized patients have a serious adverse drug reaction with a fatality rate of 0.32%.2 If these estimates are correct, then there are more than 2,216,000 serious ADRs in hospitalized patients, causing over 106,000 deaths annually. – FDA
If you recall from the earlier quotation, the estimate was 180,000 ADR deaths per year, but that was back in 2008. That was when the US population was 10% less. If we increase the figure by 10% we end up with 198,000 ADRs deaths per year. And drugs have gotten more dangerous since that time. However, I have no yet figured out how to estimate how much more dangerous.
The mortality from ADRs must be estimated, because hospitals nortiously will not add ADRs on death certificates as is shown in the following quotation.
During 1995, 206 deaths were attributed to adverse drug reactions on death certificates in the United States, whereas MedWatch tabulated 6,894 fatalities. The proportions of men and women were similar, and the majority of deaths involved persons 60 years of age and older, in both data sets. The rankings of drug categories associated with adverse drug reactions differed in the two data sets. – NCBI
And this quote…
The findings are a surprise, though, because deaths from adverse drug reactions often go unrecognized. Even when the cause is known to be a drug, that is rarely recorded on the death certificate, Dr. Pomeranz said; a certificate might list a stomach hemorrhage as the cause of death, without mentioning the drug that brought it on.
No comprehensive records are kept of medication-related deaths, Dr. Pomeranz said. Doctors and hospitals rarely report such deaths. Health care officials are not required even to keep track of them,
”Do you want to guess how many death certificates in 1994 showed deaths due to drug reactions?” Dr. Pomeranz asked. ”It was 156.”
The Food and Drug Administration, which asks doctors to report adverse drug reactions, received notice of only 3,500 such deaths in 1994. – NYT
This illustrates that deaths from drug interactions have been hidden by the pharmaceutical controlled medical system for many decades. It was only when estimates started being developed rather than relying on death certificates that more accurate estimates of ADRs were developed.
Enter Vioxx
A single pharmaceutical, Vioxx, is responsible for an estimated 500,000 deaths. This is an estimate is far below what is commonly reported in the media (around 55,000 deaths) and is explained in the following quotation.
The headline of the short article that ran in the April 19, 2005 edition of USA Today was typical: “USA Records Largest Drop in Annual Deaths in at Least 60 Years.” During that one year, American deaths had fallen by 50,000 despite the growth in both the size and the age of the nation’s population. Government health experts were quoted as being greatly “surprised” and “scratching [their] heads” over this strange anomaly, which was led by a sharp drop in fatal heart attacks.
A cursory examination of the most recent 15 years worth of national mortality data provided on the Centers for Disease Control and Prevention website offers some intriguing clues to this mystery. We find the largest rise in American mortality rates occurred in 1999, the year Vioxx was introduced, while the largest drop occurred in 2004, the year it was withdrawn. Vioxx was almost entirely marketed to the elderly, and these substantial changes in national death-rate were completely concentrated within the 65-plus population. The FDA studies had proven that use of Vioxx led to deaths from cardiovascular diseases such as heart attacks and strokes, and these were exactly the factors driving the changes in national mortality rates.
The impact of these shifts was not small. After a decade of remaining roughly constant, the overall American death rate began a substantial decline in 2004, soon falling by approximately 5 percent, despite the continued aging of the population. This drop corresponds to roughly 100,000 fewer deaths per year.
Perhaps 500,000 or more premature American deaths may have resulted from Vioxx, a figure substantially larger than the 3,468 deaths of named individuals acknowledged by Merck during the settlement of its lawsuit. And almost no one among our political or media elites seems to know or care about this possibility. – Unz
This means that for five years, the often referred to death from pharmaceuticals reactions was not the standard 198,000 pear year, but 100,000 more, or 298,000 per year.
These deaths were hidden as heart attacks and not recorded as drug reactions. It is true that the Vioxx deaths were concentrated in the 65+ age group, or in people that had far shorter future lifespans than the general population.
Once Vioxx was removed from the market (and this was not demanded by the FDA) the ADRs deaths declined significantly.
How Do Adverse Drug Reactions Deaths Compare Between Pharmaceuticals and Supplements?
It is extremely difficult to find deaths related to nutrition supplements.
Therefore, once the topic changes from hospitalizations to deaths, supplements and pharmaceuticals become incomparable. There is simply no math that can be performed that can make supplements a rational or ridiculously small fraction of the risks from pharmaceuticals.
How Do Adverse Drug Reaction Injuries Overall Compare Between Pharmaceuticals and Supplements?
In the previous quotation on those injured from drug ADRs, the estimate is over a million. This is the quote from the CDC.
Adverse drug events cause approximately 1.3 million emergency department visits each year. – CDC
However, we know that any part of the medical establishment will underestimate the number of ADRs. This is also contradicted by another area of analysis I performed, which was the number of ADRs from just the covid vaccines.
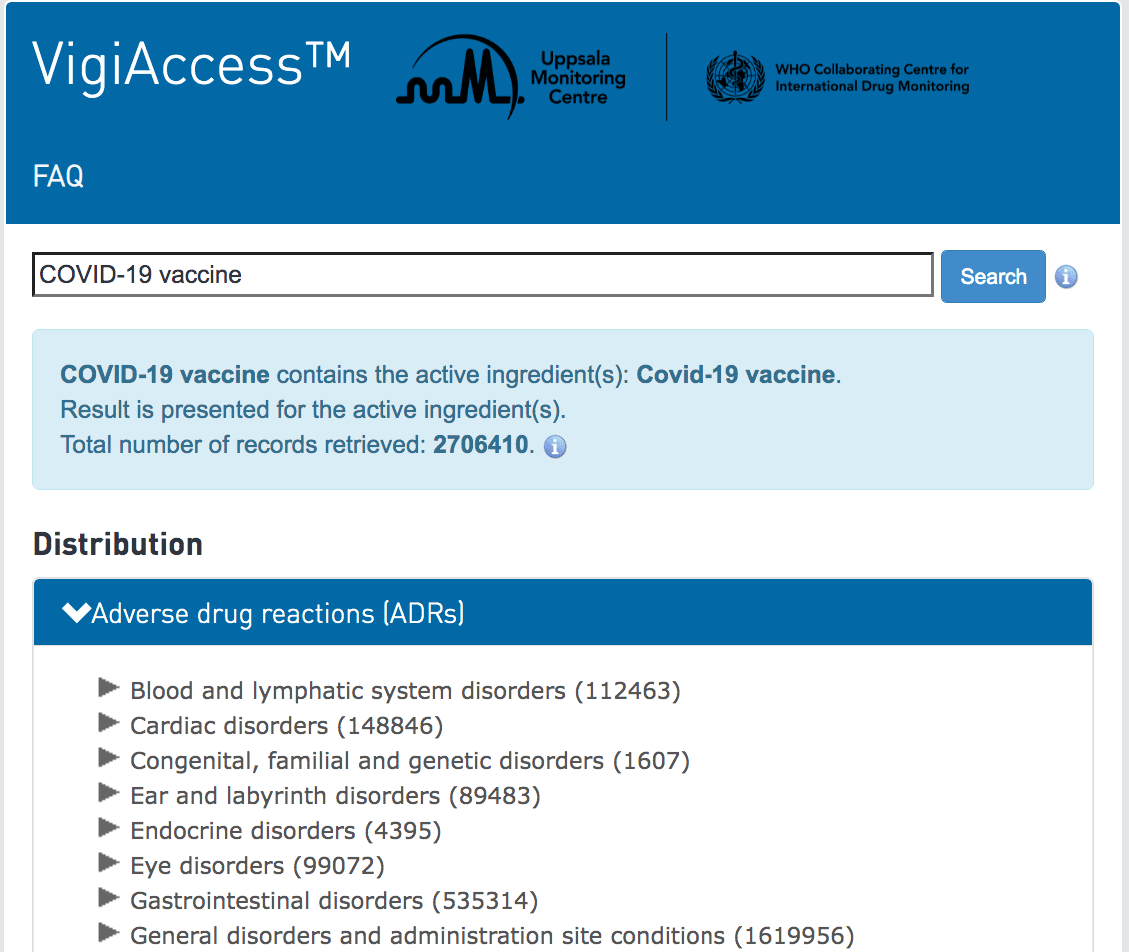
When I checked the WHO’s adverse reaction website, I found that the covid vaccines by themselves had recorded around 2.7 million adverse reactions in early December of 2021. This is over multiple years, as the vaccines were introduced not in 2021, but in 2020.

I checked around a week later and found another 100,000 adverse reactions had been added. Now, this database is global, so not just for the US.
And these are not just minor ADRs. It is now clear that the covid vaccines produce serious ADRs. The covid vaccines can make internal organs the targets of the immune system. And they are sure to cause a number of auto-immune diseases for years to come. However, when these ADRs hit in the future, they will not be attributed to the covid vaccines, but will just pop up as a new health condition. And this is the problem with thinking of ADRs that don’t lead to death as simply minor. When health authorities compare ADRs to the benefits that pharmaceuticals provide, they tend to state that pharmaceuticals save millions of lives per year. However, they don’t mention that most pharmaceuticals do not save lives. Statins do not save lives. Viagra does not save lives. Flowmax helps you pee, it does not save anyone’s life. The framing is not correct.
On Drug Adverse Reaction Underreporting
It is generally acknowledged that the reporting of adverse reactions into VAERS is very low versus the actual number of ADR. This is explained in the following quotation.
The U.S. Vaccine Adverse Event Reporting System (VAERS) is among the best adverse event data collection systems in the world, but it’s antiquated and difficult to use.
The estimated underreporting factor for COVID jab injuries in VAERS is between 31 and 100, so the actual death toll in the U.S. could be anywhere from 278,500 to 898,600.
VAERS data are being deleted without explanation. Each week, about 100 or so reports are routinely deleted, so there are now thousands of inexplicably missing reports.
VAERS, despite flaws and drawbacks, is one of the greatest tools we have to evaluate vaccine safety. It was implemented as a consequence of the 1986 National Childhood Vaccine Injury Act. While vaccine companies were given blanket immunity against liability for adverse reactions under this law, VAERS was created to collect injury reports in a centralized database so that the post-marketing safety of childhood vaccines could be monitored.
It takes on average 30 minutes to fill out a report, and the system is set up in such a way that you cannot save anything until you get to the very end.
“This probably frustrates enough people that they don’t start again,” Rose says. Indeed, the cumbersomeness of the website itself has often been cited as a reason for why doctors don’t report adverse events. Doctors don’t have the time to do it, and most patients don’t know they can file on their own. As noted by Rose: While the U.S. Food and Drug Administration and Centers for Disease Control and Prevention outrageously deny that a single death can be attributed to the COVID jabs, it’s simply impossible to discount 19,532 deaths5 (8,986 in the U.S. territories alone6) reported as of November 26, 2021. As noted by Rose: As mentioned, Kirsch has calculated an underreporting factor for post COVID jab events of 41, which is likely quite conservative. Rose’s calculation is even more conservative than that. She explains:
“Steve [Kirsch] and I are good friends. We’ve been working very closely on all of this stuff for a long time. His underreporting factor is 41. He estimated that based on a peer-reviewed publication that estimated anaphylaxis numbers, so he used anaphylaxis as a proxy for death.
What that means is that when you hear us say these numbers, you have to multiply them by 41, if you want to go with Steve’s estimate, or 31, in the case of mine. Mine is the most conservative estimate. I took Pfizer’s Phase 3 clinical trial data that they presented to the FDA. – Mercola
If the factor of 41 is used to account for underreporting, we could take 2.8 M x 41 to obtain 110 M. This means that even when we cut out just the US number, the total ADR from covid would be far higher than the estimates from the CDC. And this is just the ADRs from the vaccines.
Other estimates for ADRs I have seen have been higher, at around 2.1 M. However this is far too low.
What all of this means is that the estimates normally provided on ADRs are absurdly too low. It is in the multi-millions. And the typical ADR from a pharmaceutical is far more serious than an ADR from a supplement. So both the incidence and the severity of the adverse reactions between pharmaceuticals and supplements is not really comparable.
How the Safety Pharmaceuticals Has Declined
It seems like adverse drug reactions are far more the place to focus attention. Adverse events from drugs are increasing, as is explained in the following video.
Observe the graph which is shown at the 6-minute mark that illustrates the constant rise in adverse events.
This quote from The Harvard Ethics website covers the declining safety of drugs.
But as far as we can tell (very little research is funded on prescription drugs as a health risk compared to less deadly risks like diabetes or Alzheimer’s disease), millions who take new, patented drugs experience only modest benefits over established drugs. Only a small percent of new drugs provide significant advantages for patients to offset these risks of harm. Independent reviews over the past 35 years have found that only 11 to 15 percent of newly approved drugs have significant clinical advantages over existing, better-known drugs.
Closely coordinated in the trial-journal pipeline, pharmaceutical companies retain teams of statisticians, science editors, and science writers to select which results will go into the medical literature and which will not. They switch end points and other details in the data submitted to the FDA so that physicians read twice-biased medical articles that understate risks of harm and overstate benefits. Negative results are much less likely to be published than positive results, and companies publish positive results more than once, a further bias that distorts clinical practice and guidelines as well the medical knowledge that underlies it. – Harvard Ethics
Yes.
Because the FDA has been captured by pharmaceutical companies, very unsafe drugs are getting approved so that FDA employees can receive off-the-books financial benefits from pharmaceutical companies. The American Cancer Society is part of this corruption, as they can be viewed as an extension of the pharmaceutical and radiological industries.
This video explains the corruption on the part of the FDA. The FDA claims it is incapable of stopping opioids from being prescribed because they “lack the manpower” in an organization with 15,000 people that has the power to remove the approval of any drug at any time.
In response to drug disasters like Vioxx, which experts say caused about 120,000 traumatic cardiovascular events and 40,000 deaths, Congress and the FDA have set up monitoring and safety systems.
But a review of results so far found little evidence they are identifying serious risks or altering prescribing practices. – Harvard Ethics
Amazingly, after Vioxx was proven to be a very dangerous drug that was so deadly it significantly altered the US death rate, the drug did not have its approval pulled by the FDA. This is explained by the FDA’s website.
Did FDA require this action?
No, Merck made this decision independent of input from FDA. The Agency has not had an opportunity to review the data from the study that was stopped in the depth that Merck has, but agrees with the company that there appear to be significant safety concerns for patients, particularly those taking the drug chronically.
FDA plans to work closely with Merck to coordinate the withdrawal of this product from the US market.
Merck’s decision to withdraw Vioxx from the market is based on new data from a trial called the APPROVe [ Adenomatous Polyp Prevention on VIOXX] trial. In the APPROVe trial, Vioxx was compared to placebo (sugar-pill). The purpose of the trial was to see if Vioxx 25 mg was effective in preventing the recurrence of colon polyps. This trial was stopped early because there was an increased risk for serious cardiovascular events, such as heart attacks and strokes, first observed after 18 months of continuous treatment with Vioxx compared with placebo. – FDA
This article was written by the FDA in 2004, in response to Merck pulling the drug from the market. However, an article in the Lancet disputes the FDA’s explanation of the events.
These findings also come in the wake of new disclosures that suggest Merck was indeed fully aware of Vioxx’s potential risks by 2000. Investigations by the Wall Street Journal2 have revealed e-mails that confirm Merck executives’ knowledge of their drug’s adverse cardiovascular profile—the risk was “clearly there”, according to one senior researcher. Merck’s marketing literature included a document intended for its sales representatives which discussed how to respond to questions about Vioxx—it was labelled “Dodge Ball Vioxx”.
Worse still, the FDA’s Office of Drug Safety co-exists in the same centre—the Centre for Drug Evaluation and Research (CDER)—as the Office of New Drugs, the part of the agency that works most closely with industry to license new medicines. Once a licensing approval has been made it is naturally in CDER’s own interests to stand by its original decision. CDER’s reputation would be damaged if its licensing judgments were constantly challenged by its own staff. This understandable but dangerous tendency to discourage dissent makes the Office of Drug Safety, which sits lower in the hierarchy of CDER than the Office of New Drugs, weak and ineffective. The inherent precedence that licensing of new drugs takes over safety evaluation is a serious flaw in FDA’s complex regulatory structure.
In the case of Vioxx, FDA was urged to mandate further clinical safety testing after a 2001 analysis suggested a “clear-cut excess number of myocardial infarctions”.3 It did not do so. This refusal to engage with an issue of grave clinical concern illustrates the agency’s in-built paralysis, a predicament that has to be addressed through fundamental organisational reform.
On Nov 2, 2004, the FDA tried to shore up its tarnished reputation by posting on its website an early version of a recently completed observational study into the safety of Vioxx. The report comes with a warning that it has “not been fully evaluated by the FDA and may not reflect the official views of the agency”.
This study is presently under review at The Lancet. It is unclear why the FDA could not have waited for the fully evaluated report to have been scrutinised, revised, and published according to the norms of scientific peer review. Bypassing independent peer review smacks of panic in the FDA, which is under intense reputational pressure. And yet its decision to try to undermine the integrity of this work again shows that the agency’s senior management is more concerned with external appearance than rigorous science.
The licensing of Vioxx and its continued use in the face of unambiguous evidence of harm have been public-health catastrophes. This controversy will not end with the drug’s withdrawal. Merck’s likely litigation bill is put at between US$10 and $15 billion.
For with Vioxx, Merck and the FDA acted out of ruthless, short-sighted, and irresponsible self-interest.
This is the article authors calling out not only Merck but also the FDA for completely lying. Both entities knew the cardiovascular dangers and went ahead with the drug anyway.
In the next quote, the FDA tries to deny that its expedited review process is causing more dangerous drugs to be approved.
Is FDA’s expedited review process putting riskier drugs on the market?
No. Vioxx received a six-month priority review because the drug potentially provided a significant therapeutic advantage over existing approved drugs due to fewer gastrointestinal side effects, including bleeding. A product undergoing a priority review is held to the same rigorous standards for safety, efficacy, and quality that FDA expects from all drugs submitted for approval. – FDA